Key points
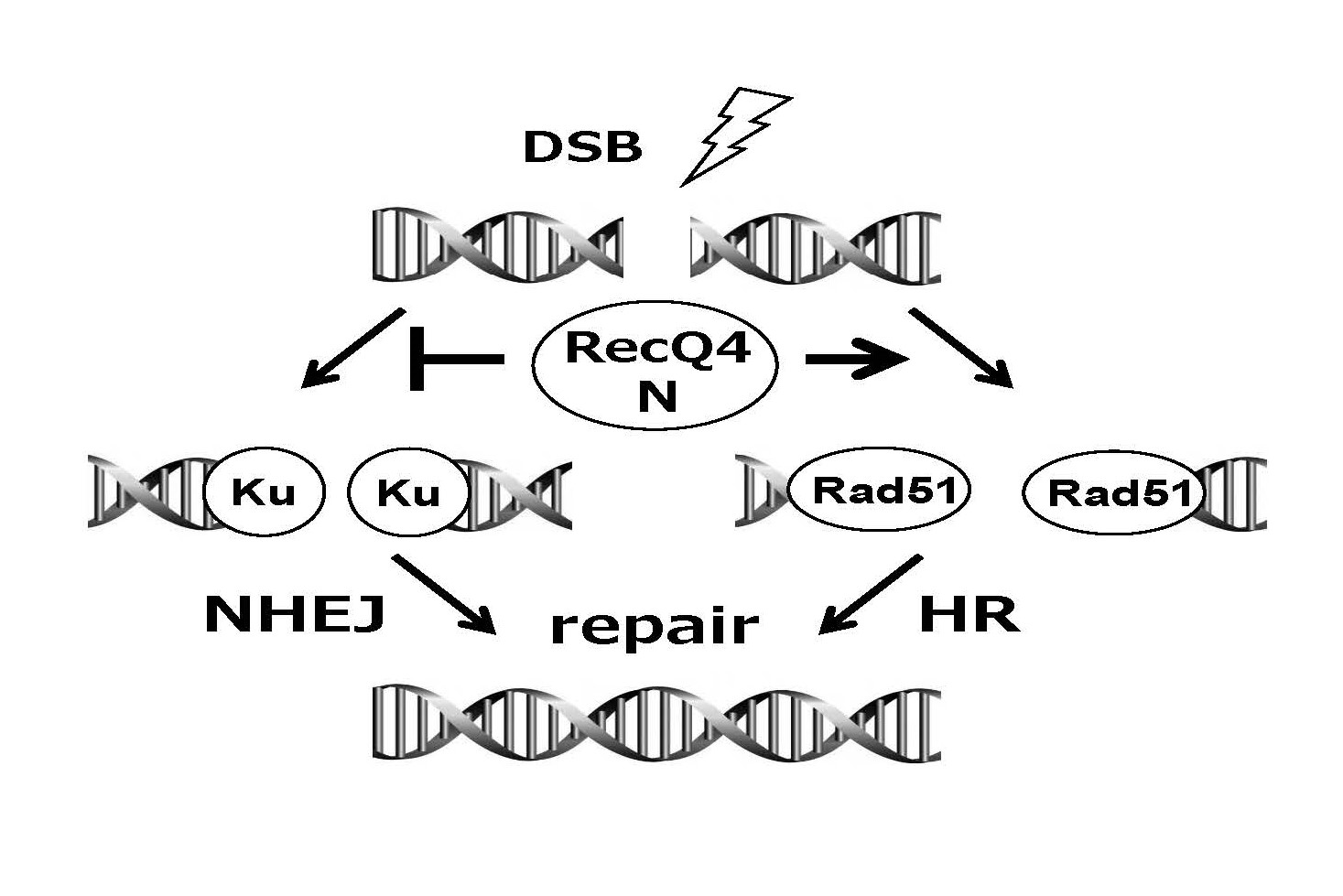
RecQ4 was implicated as the causative agent of Rothmund-Thomson syndrome. The authors demonstrated that the N-terminal fragment of RecQ4 was shown to inhibit non-homologous end-joining repair, which is important in DNA double-strand break repair. Further analyses revealed that RecQ4 inhibits the binding of Ku70, an essential protein for non-homologous end-joining repair, to DNA double-strand breaks, and the inhibition is dependent on the enzymatic activity of ATM, a serine/threonine kinase involved in homologous recombination repair.
The findings clarify the role of RecQ4 in DNA double-strand break repair and the pathogenesis of the Rothmund-Thomson syndrome. The findings also enhance our molecular understanding of aging and carcinogenesis.
Summary
DNA double-strand breaks (DSBs) are induced in response to radiation or anticancer drugs. DSBs are the most serious form of DNA damage. Repair of DSBs mainly involves one of two repair pathways: non-homologous end-joining repair and homologous recombination repair. Cells appear to have the ability to select between these repair pathways depending on the situation. The pathway selection mechanism has been studied more intensively in recent years. RecQ4 is mutated in the Rothmund-Thomson syndrome, a disease characterized by premature aging and high risk of carcinogenicity. This protein functions in the DSB repair pathway, but the details of its function are unclear. The research group led by Professor Shusuke Tada and Lecturer Takashi Tsuyama at the Department of Molecular Biology, Faculty of Pharmaceutical Sciences, Toho University, established an experimental system that can reproduce the DSB repair system in Xenopus egg extracts. Experimental results suggested the involvement of RecQ4 in the pathway selection of DSB repair. The results of this study help elucidate the role of RecQ4 in DNA repair and will further our understanding of the molecular underpinnings of the pathogenesis of Rothmund-Thomson syndrome, aging, and cancer.